🦠Microbiology Unit 14 Review
14.3 Mechanisms of Antibacterial Drugs
14.3 Mechanisms of Antibacterial Drugs
Unit & Topic Study Guides
How We See the Invisible World
The Cell
Prokaryotic Diversity
The Eukaryotes of Microbiology
Acellular Pathogens
Microbial Biochemistry
Microbial Metabolism
Microbial Growth
Biochemistry of the Genome
Mechanisms of Microbial Genetics
Modern Applications of Microbial Genetics
Control of Microbial Growth
Antimicrobial Drugs
Microbial Mechanisms of Pathogenicity
Disease and Epidemiology
Innate Nonspecific Host Defenses
Adaptive Specific Host Defenses
Diseases of the Immune System
Laboratory Analysis of the Immune Response
Skin and Eye Infections
Respiratory System Infections
Urogenital System Infections
Digestive System Infections
Circulatory and Lymphatic System Infections
Selective Targeting and Mechanisms of Action
Antibacterial drugs work by exploiting key differences between bacterial and human cells. Bacteria have unique structures (like peptidoglycan cell walls and 70S ribosomes) and metabolic pathways (like folic acid synthesis) that human cells lack. By targeting these bacterial-specific features, antibiotics can kill or inhibit bacteria while causing minimal harm to the host.
This concept of selective toxicity is the foundation of antibiotic therapy. The more selectively a drug targets a bacterial structure, the fewer side effects it tends to cause.
How selectivity works in practice
Bacterial cells offer several targets that differ from human cells:
- Cell wall enzymes: Transpeptidases and penicillin-binding proteins (PBPs) build peptidoglycan, a structure human cells don't have at all.
- Ribosomes: Bacterial ribosomes are 70S (made of 30S + 50S subunits), while human ribosomes are 80S (40S + 60S subunits). The structural differences at specific binding sites allow drugs to block bacterial translation without disrupting ours.
- Folic acid synthesis: Bacteria must synthesize their own folic acid. Humans get folate from food, so drugs that block this pathway in bacteria have no equivalent target in human cells.
- DNA-handling enzymes: Bacterial DNA gyrase and topoisomerase IV differ enough from human topoisomerases that certain drugs can inhibit the bacterial versions selectively.
The goal in drug design is high affinity for the bacterial target and low affinity for anything in human cells. When a drug also interacts with human structures, that's where side effects come from.
Mechanisms of antibacterial drug classes
Each class of antibiotic disrupts a different essential process in the bacterial cell. Here's how the major classes work:
Cell wall synthesis inhibitors
These drugs prevent bacteria from building or maintaining their peptidoglycan cell wall. Without a functional wall, the bacterium can't withstand osmotic pressure and lyses. These are bactericidal (they kill bacteria, not just stop growth).
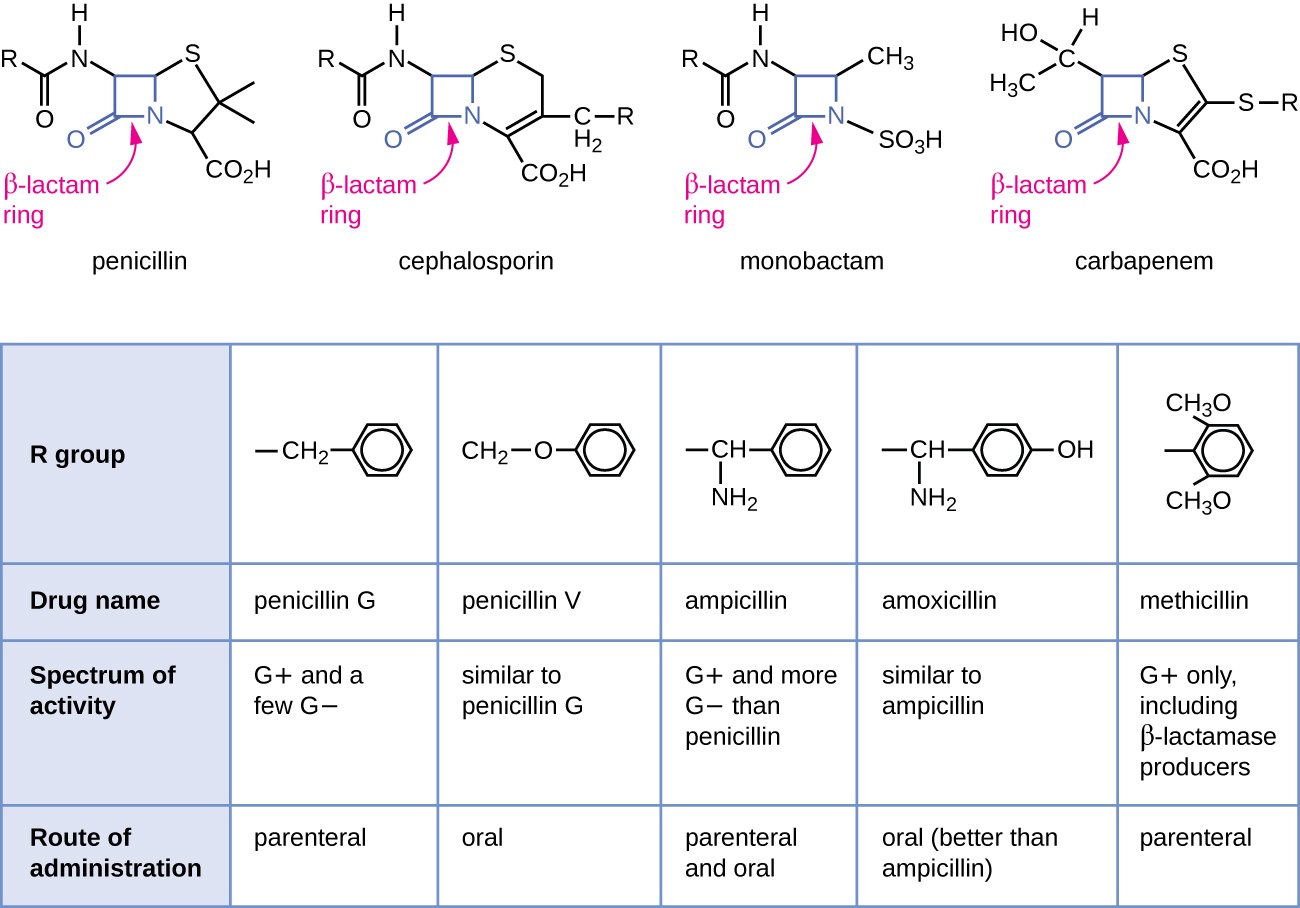
- Beta-lactams (penicillins, cephalosporins, carbapenems, monobactams)
- Bind to and inhibit transpeptidase enzymes (PBPs), which are responsible for cross-linking peptidoglycan strands
- Without cross-links, the cell wall weakens, and the cell undergoes osmotic lysis
- Glycopeptides (vancomycin)
- Bind to the D-alanine-D-alanine terminal residues on peptidoglycan precursors
- This physically blocks the precursors from being incorporated into the growing cell wall, preventing both synthesis and cross-linking
A useful distinction: beta-lactams inhibit the enzyme that does the cross-linking, while vancomycin binds the substrate (the peptidoglycan precursor itself). Different strategy, same result.
Protein synthesis inhibitors
These drugs bind to bacterial ribosomal subunits and disrupt translation at various stages. Because they target the 30S or 50S subunit of the bacterial 70S ribosome, they generally spare human 80S ribosomes.
Drugs targeting the 30S subunit:
- Aminoglycosides (gentamicin, tobramycin)
- Bind the 30S subunit and cause misreading of mRNA, so wrong amino acids get inserted
- Also cause premature termination of translation
- The result is nonfunctional, misfolded proteins; bactericidal
- Tetracyclines
- Bind the 30S subunit and block aminoacyl-tRNA from attaching to the ribosome's A site
- Translation stalls because no new amino acids can be added; bacteriostatic
Drugs targeting the 50S subunit:
- Macrolides (erythromycin, azithromycin), lincosamides (clindamycin), and streptogramins
- Bind the 50S subunit and inhibit translocation or peptidyl transferase activity
- The polypeptide chain can't elongate properly; bacteriostatic (though bactericidal at high concentrations for some)
- Oxazolidinones (linezolid)
- Bind the 50S subunit and prevent formation of the 70S initiation complex
- Translation never even starts; bacteriostatic
Nucleic acid synthesis inhibitors
These drugs interfere with DNA replication, transcription, or the production of nucleotide precursors.
- Fluoroquinolones (ciprofloxacin, levofloxacin)
- Inhibit DNA gyrase and topoisomerase IV, enzymes bacteria need to supercoil DNA and separate daughter strands during replication
- DNA replication stalls and the chromosome fragments; bactericidal
- Rifamycins (rifampin)
- Bind to bacterial RNA polymerase and block transcription initiation
- Without mRNA, the cell can't make proteins; bactericidal
- Rifampin targets the beta subunit of RNA polymerase, which differs structurally from human RNA polymerases
Antimetabolites (folic acid pathway inhibitors)
- Sulfonamides inhibit dihydropteroate synthase, the first enzyme in the bacterial folic acid synthesis pathway
- Trimethoprim inhibits dihydrofolate reductase, the next enzyme in the same pathway
- Together, they create a sequential blockade: two steps in the same pathway are shut down, which is why sulfonamides and trimethoprim are often prescribed in combination (e.g., TMP-SMX, also called co-trimoxazole)
- Without folate, bacteria can't synthesize nucleotides for DNA replication; bacteriostatic individually, bactericidal in combination
Spectrum of Activity
The spectrum of activity describes the range of bacterial species an antibiotic is effective against. This matters for choosing the right drug and for understanding the trade-offs of each approach.
Broad-spectrum vs. narrow-spectrum antibiotics
Broad-spectrum drugs (e.g., tetracyclines, fluoroquinolones, carbapenems) are effective against both Gram-positive and Gram-negative bacteria.
- Useful for empiric therapy, when you need to start treatment before lab results identify the pathogen
- The trade-off: they also kill off normal microbiota, which can lead to:
- GI disturbances (diarrhea, nausea)
- Superinfections from opportunistic organisms like Clostridioides difficile, which thrives when competing bacteria are wiped out
- Greater selection pressure for antibiotic resistance across many bacterial species
Narrow-spectrum drugs (e.g., vancomycin for Gram-positives, aztreonam for Gram-negatives) target a limited range of bacteria.
- Cause less collateral damage to normal microbiota
- Put less selection pressure on non-target bacteria, helping preserve antibiotic effectiveness
- The trade-off: they require accurate identification of the pathogen. If the diagnosis is wrong, the drug may not work. Polymicrobial infections may need multiple narrow-spectrum agents.
The general principle: use the narrowest spectrum that covers the pathogen. Broad-spectrum drugs are a safety net when you don't yet know what you're treating, but narrowing therapy once culture results come back is standard practice.

Pharmacokinetics and Pharmacodynamics
These two concepts describe different sides of the drug-body relationship and are essential for understanding how dosing works.
Pharmacokinetics (PK) is what the body does to the drug. It covers four processes, often abbreviated ADME:
- Absorption: How the drug enters the bloodstream (oral, IV, etc.)
- Distribution: How the drug travels to tissues and the infection site
- Metabolism: How the body chemically modifies the drug (mostly in the liver)
- Excretion: How the drug is eliminated (mostly through the kidneys)
PK determines how much drug actually reaches the bacteria and for how long. This directly shapes dosing schedules and route of administration.
Pharmacodynamics (PD) is what the drug does to the body (and to the bacteria). It includes:
- The relationship between drug concentration and antimicrobial effect
- The minimum inhibitory concentration (MIC), which is the lowest drug concentration that prevents visible bacterial growth in vitro
- Whether a drug's killing depends on peak concentration (concentration-dependent, like aminoglycosides) or time spent above the MIC (time-dependent, like beta-lactams)
Understanding PK/PD together helps clinicians design dosing regimens that maximize bacterial killing while minimizing toxicity and resistance development.
Antibiotic resistance
Bacteria can develop resistance through spontaneous genetic mutations or by acquiring resistance genes from other bacteria (via plasmids, transposons, or transformation). Resistance mechanisms include:
- Producing enzymes that destroy the drug (e.g., beta-lactamases that break down penicillins)
- Altering the drug's target so it no longer binds effectively
- Reducing drug uptake or increasing efflux (pumping the drug out of the cell)
- Bypassing the inhibited pathway entirely
Resistance is an ongoing clinical challenge. It's why susceptibility testing, antibiotic stewardship (using antibiotics only when needed, and choosing the narrowest effective spectrum), and the development of new drugs all remain critical priorities.