Solution-Phase Reaction Kinetics
Solution-phase reaction kinetics examines how reactions behave in liquid environments, where the solvent isn't just a passive backdrop. The solvent's polarity, viscosity, and ionic content all shape how fast reactions proceed and which mechanisms dominate. This matters because most real-world chemistry (biological, industrial, pharmaceutical) happens in solution, not in the gas phase.
This section covers steady-state approximations for deriving rate laws, how solvents influence rates, the role of ionic strength, and how the Arrhenius equation applies to solution-phase reactions.
Steady-State Approximation for Rate Laws
Many reaction mechanisms involve reactive intermediates that form in one step and get consumed in the next. The steady-state approximation (SSA) assumes that after a brief induction period, the concentration of these intermediates stays roughly constant over time. That is, the rate at which the intermediate forms equals the rate at which it's consumed.
This works because intermediates are typically highly reactive and short-lived. They never accumulate to significant concentrations, so .
To apply the steady-state approximation:
- Identify the reactive intermediate(s) in the proposed mechanism. These are species that appear in elementary steps but not in the overall balanced equation (e.g., a carbocation in an SN1 mechanism).
- Write rate expressions for every elementary step that forms or consumes each intermediate.
- Set the net rate of change of the intermediate to zero and solve algebraically for its concentration in terms of reactant concentrations and rate constants.
- Substitute that expression into the rate equation for the product-forming step to get the overall rate law.
The resulting rate law can then be compared to experimental data to validate or reject the proposed mechanism.

Solvent Effects on Reaction Rates
In solution, the solvent interacts with reactants, products, intermediates, and transition states. These interactions can dramatically speed up or slow down a reaction.
- Polarity: Polar solvents stabilize charged or highly polar intermediates and transition states. For example, SN1 reactions speed up in polar protic solvents (like water) because the solvent stabilizes the carbocation intermediate. Conversely, SN2 reactions often proceed faster in polar aprotic solvents (like DMSO) because these solvents don't strongly solvate the nucleophile, leaving it more reactive.
- Viscosity: Reactant molecules must diffuse through the solvent to encounter each other. In highly viscous solvents like glycerol, diffusion slows down, which reduces the rate of bimolecular reactions. This is especially relevant for diffusion-controlled reactions, where the rate-limiting step is simply how fast reactants can find each other.
- Dielectric constant: A solvent's dielectric constant reflects its ability to reduce electrostatic interactions between charges. High-dielectric solvents (like water, ) stabilize ions and ion pairs more effectively than low-dielectric solvents (like diethyl ether, ).
The Hughes-Ingold rules provide a systematic way to predict solvent effects. The key idea: if the transition state is more charge-separated than the reactants, increasing solvent polarity speeds up the reaction. If the transition state is less charge-separated, increasing polarity slows it down.
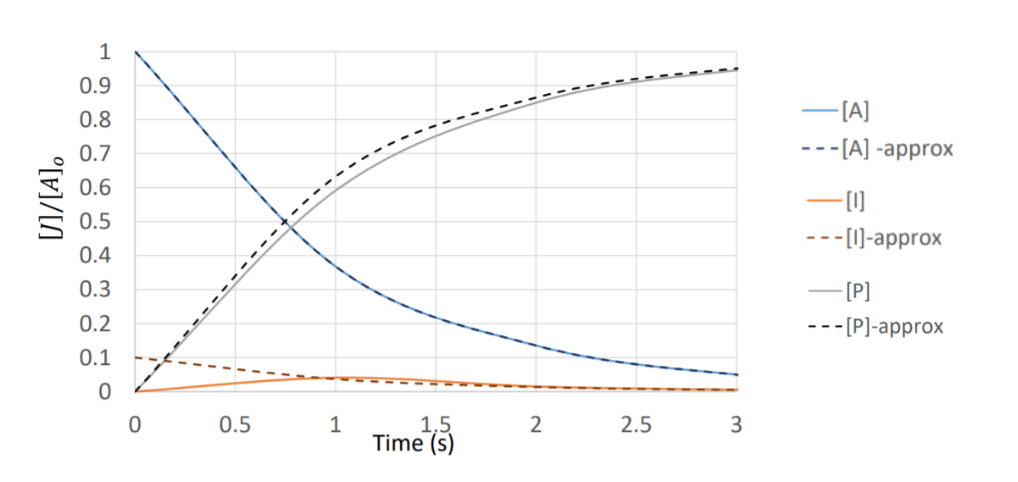
Ionic Strength in Reaction Kinetics
When reactions involve ionic species in solution, the surrounding ions influence the effective concentrations (activities) of the reactants. Ionic strength () quantifies the total ion concentration in solution:
where is the molar concentration and is the charge of each ion.
Debye-Hückel theory explains why ionic strength matters: surrounding ions form an "ionic atmosphere" around each charged reactant, partially screening its charge. As ionic strength increases, activity coefficients decrease, meaning the effective concentration of each ion deviates more from its actual concentration.
The Brønsted-Bjerrum equation connects ionic strength directly to the observed rate constant:
- = observed rate constant at ionic strength
- = rate constant at zero ionic strength (infinite dilution)
- = a constant that depends on the solvent and temperature (approximately 0.509 in water at 25°C)
- , = charges of the two reacting species
- = ionic strength
The product determines the direction of the effect. If both reactants carry the same sign of charge (both positive or both negative), , and increasing ionic strength increases the rate. If the reactants carry opposite charges, , and increasing ionic strength decreases the rate. This is called the primary kinetic salt effect.
Activation Energy of Solution Reactions
The Arrhenius equation relates the rate constant to temperature and the energy barrier for reaction:
- = rate constant
- = pre-exponential (frequency) factor, related to how often molecules collide with the correct orientation
- = activation energy (the minimum energy needed to reach the transition state)
- = gas constant (8.314 J/mol·K)
- = absolute temperature in Kelvin
To determine and experimentally:
-
Measure the rate constant at several different temperatures.
-
Take the natural log of the Arrhenius equation to get the linear form:
-
Plot on the y-axis vs. on the x-axis. This Arrhenius plot should give a straight line.
-
Extract the parameters:
- Slope = , so
- y-intercept = , so
A large means the reaction is very sensitive to temperature changes, while a small means temperature has a weaker effect. The pre-exponential factor captures the frequency and orientation requirements of productive collisions.
In solution, values reflect not just the intrinsic bond-breaking/forming process but also the energy cost of reorganizing solvent molecules around the transition state. For instance, SN1 reactions in polar solvents may have lower effective activation energies than in nonpolar solvents because the solvent stabilizes the charge-separated transition state.