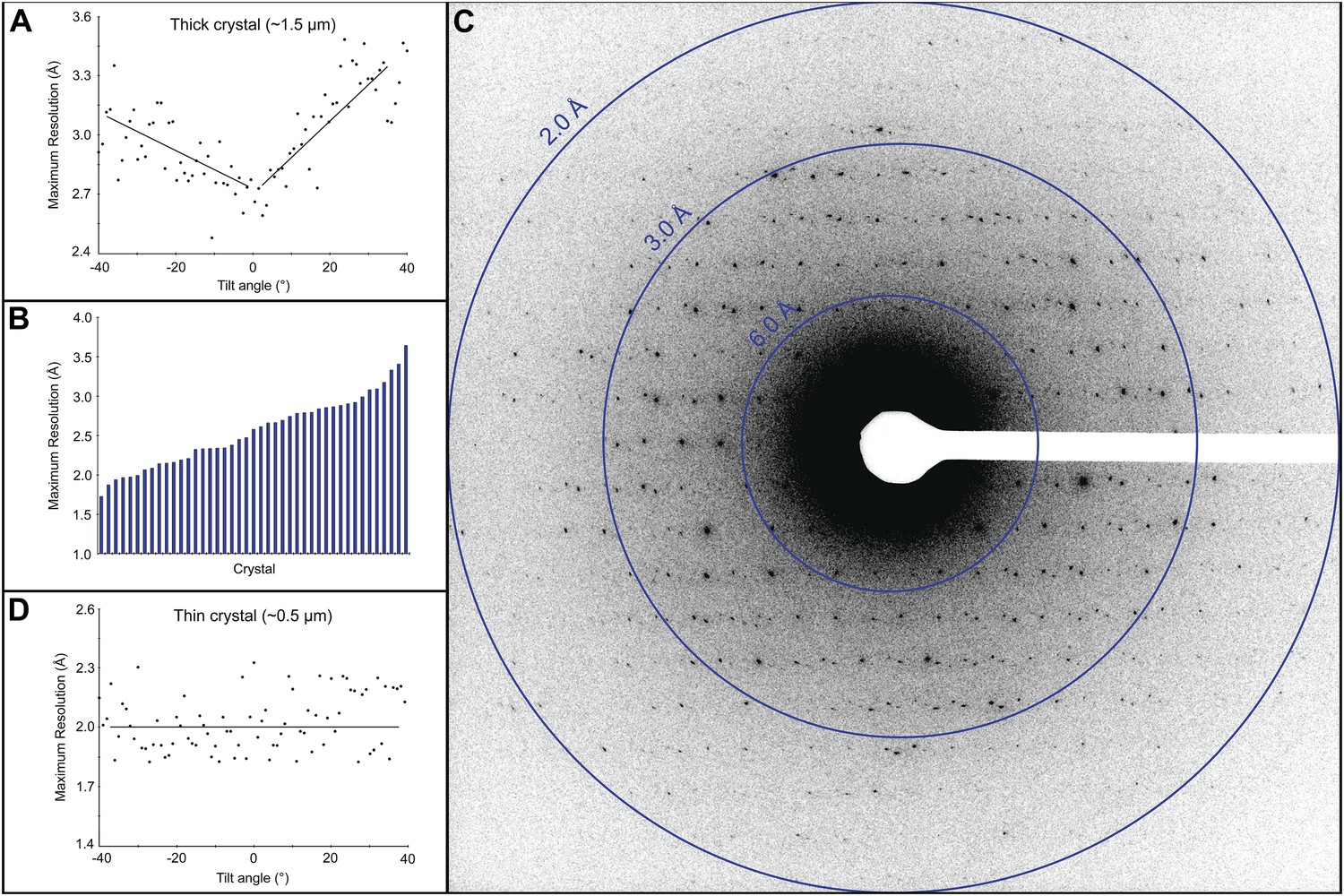
Structure solution strategies are crucial for unraveling crystal structures from diffraction data. These methods tackle the phase problem, using statistical relationships, heavy atoms, or known structures to determine atomic positions.
Advanced techniques like charge flipping and experimental phasing expand our toolkit. By interpreting electron density maps, we can visualize atomic arrangements and refine our structural models, bridging raw data and final crystal structures.
Structure Solution Methods
Direct Methods and Patterson Techniques
- Direct methods utilize statistical relationships between structure factors to determine phases
- Relies on atomicity principle and positivity of electron density
- Works well for small molecules with up to ~200 atoms
- Involves generating probable phase sets and evaluating their likelihood
- Uses normalized structure factors to enhance atomic resolution
- Patterson methods analyze the Patterson function to locate heavy atoms or molecular fragments
- Patterson map represents vector distances between atoms in the structure
- Peaks in Patterson map correspond to interatomic vectors
- Particularly useful for structures with a few heavy atoms (uranium)
- Can determine heavy atom positions without phase information
Advanced Structure Solution Approaches
- Charge flipping algorithm iteratively modifies electron density to solve structures
- Alternates between real and reciprocal space calculations
- Flips the sign of weak electron density regions in each cycle
- Does not require prior knowledge of chemical composition
- Effective for solving structures of complex materials (quasicrystals)
- Molecular replacement utilizes known structures to solve new, related structures
- Requires a homologous model structure as a starting point
- Involves rotation and translation searches to position the model in the unit cell
- Particularly useful for protein crystallography where similar structures often exist
- Can significantly speed up structure solution process for macromolecules
Experimental Phasing Techniques
Isomorphous Replacement Methods
- Isomorphous replacement introduces heavy atoms to the crystal without changing its structure
- Compares diffraction patterns of native and heavy atom-containing crystals
- Single isomorphous replacement (SIR) uses one heavy atom derivative
- Multiple isomorphous replacement (MIR) uses multiple heavy atom derivatives for improved accuracy
- Calculates phase information from intensity differences between native and derivative crystals
- Heavy atom method exploits the strong scattering of heavy atoms to determine phases
- Locates heavy atom positions using Patterson or direct methods
- Uses heavy atom positions to estimate initial phases for the entire structure
- Particularly effective for large biological molecules (proteins)
Anomalous Dispersion Techniques
- Anomalous dispersion arises from absorption of X-rays by certain atoms
- Causes breakdown of Friedel's law, where
- Single-wavelength anomalous dispersion (SAD) uses data collected at one wavelength
- Multi-wavelength anomalous dispersion (MAD) uses data from multiple wavelengths
- Often employs selenium atoms incorporated into proteins for phasing
- Combines aspects of isomorphous replacement and anomalous scattering
- Single isomorphous replacement with anomalous scattering (SIRAS)
- Multiple isomorphous replacement with anomalous scattering (MIRAS)
- Enhances phase determination accuracy by utilizing both methods
Interpreting Results
Phase Problem and Its Solutions
- Phase problem arises from the inability to directly measure phases in diffraction experiments
- Diffraction data only provides amplitudes of structure factors
- Phases are crucial for reconstructing the electron density map
- Various methods (direct, Patterson, experimental phasing) address this issue
- Phase refinement and improvement techniques enhance initial phase estimates
- Density modification methods (solvent flattening, histogram matching)
- Iterative phase improvement cycles combined with model building
- Maximum likelihood methods for phase probability estimation
Electron Density Map Analysis
- Electron density map represents the distribution of electrons in the crystal structure
- Calculated using structure factor amplitudes and estimated phases
- Different map types (Fo-Fc, 2Fo-Fc) highlight various structural features
- Interpretation of electron density maps requires expertise and often iterative refinement
- Identifies atomic positions, bond lengths, and molecular geometry
- Automated model-building software assists in initial structure interpretation
- Manual inspection and refinement crucial for accurate final structures
- Resolution of the map affects the level of detail that can be interpreted