Molecular dynamics simulations are powerful tools for studying chemical reactions at the atomic level. They use Newton's equations to track atoms' movements over time, exploring the potential energy surface that governs a system's behavior. This method offers unique insights into reaction pathways and energy barriers.
These simulations complement experimental techniques by providing detailed views of molecular interactions. They can reveal reaction mechanisms, identify intermediate states, and study conformational changes in complex biomolecules. This approach bridges the gap between theory and experiment in understanding reaction dynamics.
Molecular Dynamics Simulations
Principles and Applications
- Molecular dynamics simulations numerically solve Newton's equations of motion for a system of interacting atoms or molecules
- Provides a trajectory of atomic positions and velocities over time
- The potential energy surface (PES) describes the potential energy of a molecular system as a function of its atomic coordinates
- Governs the forces acting on the atoms and the system's dynamic behavior
- Molecular dynamics simulations can explore the PES
- Samples different regions of configuration space
- Provides insights into reaction pathways, transition states, and energy barriers
- Applications of molecular dynamics simulations in studying reaction dynamics
- Investigates the microscopic details of chemical reactions (bond breaking and formation, solvent effects)
- Identifies reaction mechanisms, intermediate states, and rate-determining steps
- Studies conformational changes and structural transitions in biomolecules (protein folding, ligand binding)
- Molecular dynamics simulations can complement experimental techniques
- Provides atomic-level insights that may be difficult to obtain experimentally
- Aids in the interpretation of experimental data
Potential Energy Surface and System Dynamics
- The potential energy surface (PES) is a multidimensional surface that represents the potential energy of a molecular system as a function of its atomic coordinates
- The shape of the PES determines the forces acting on the atoms and the system's dynamic behavior
- Minima on the PES correspond to stable configurations (reactants, products, intermediates)
- Saddle points on the PES represent transition states between stable configurations
- Molecular dynamics simulations can sample different regions of the PES
- Explores the accessible configurational space of the system
- Identifies low-energy pathways connecting reactants to products
- Estimates the free energy barriers associated with different reaction pathways
- The gradient of the PES with respect to atomic coordinates gives the forces acting on the atoms
- These forces are used to update atomic positions and velocities at each time step of the simulation
- The resulting trajectory provides a time-dependent picture of the system's evolution on the PES
Setting Up and Running Simulations
Force Fields and Interatomic Interactions
- Choosing an appropriate force field is crucial for accurately describing the interatomic interactions in the system
- Force fields define the functional form and parameters for bonded interactions (bond stretching, angle bending, torsional terms)
- Non-bonded interactions (van der Waals, electrostatic) are also included in the force field
- Common force fields for biomolecular simulations include AMBER, CHARMM, and GROMOS
- These force fields have been parameterized to reproduce experimental data (structures, energetics, dynamics)
- Polarizable force fields (AMOEBA, Drude oscillator) can capture electronic polarization effects
- Important for systems with significant charge redistribution or induced dipoles
- Reactive force fields (ReaxFF, AIREBO) can describe bond breaking and formation
- Enables the simulation of chemical reactions without predefined reaction coordinates
System Preparation and Initialization
- Initial atomic coordinates can be obtained from experimental structures
- X-ray crystallography or NMR provide high-resolution structures of biomolecules
- Molecular modeling software can generate initial structures for small molecules or systems without experimental data
- The simulation box size, shape, and density should be chosen to accurately represent the system of interest
- Periodic boundary conditions are often used to simulate bulk systems and minimize edge effects
- The box size should be large enough to avoid artificial interactions between periodic images
- Solvent molecules (water, ions) are added to the simulation box to mimic the desired environment
- Explicit solvent models (TIP3P, SPC/E) represent water molecules as discrete entities
- Implicit solvent models (Generalized Born, Poisson-Boltzmann) treat the solvent as a continuum dielectric medium
- The system is energy minimized to remove any initial bad contacts or strain
- Steepest descent or conjugate gradient methods are commonly used for energy minimization
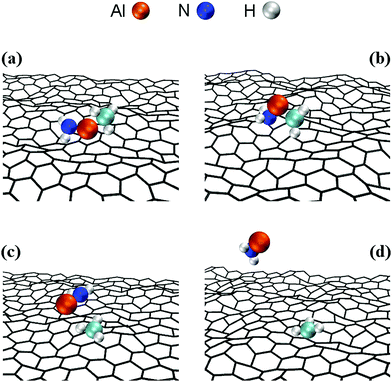
Simulation Parameters and Protocols
- Thermodynamic ensembles (microcanonical, canonical, isothermal-isobaric) are selected based on the desired control of temperature, pressure, and other thermodynamic variables
- The canonical ensemble (NVT) maintains a constant number of particles, volume, and temperature
- The isothermal-isobaric ensemble (NPT) maintains a constant number of particles, pressure, and temperature
- Integration algorithms (Verlet, leapfrog) are used to numerically integrate the equations of motion and update atomic positions and velocities at each time step
- The Verlet algorithm is time-reversible and symplectic, ensuring energy conservation
- The leapfrog algorithm is a modified Verlet algorithm that updates positions and velocities at interleaved time points
- The choice of time step depends on the fastest motions in the system (typically bond vibrations)
- Time steps are usually on the order of 1-2 fs for all-atom simulations
- Constraints (SHAKE, LINCS) can be applied to bond lengths, allowing for larger time steps
- Equilibration simulations are performed to allow the system to relax and reach a stable state
- Thermodynamic properties (temperature, pressure, energy) are monitored to ensure equilibration
- Production simulations are then run to collect data for analysis
Analyzing Simulation Results
Trajectory Analysis and Reaction Coordinates
- Trajectory analysis involves examining the time evolution of atomic positions, velocities, and energies
- Identifies key events and structural changes during the reaction
- Calculates various properties (distances, angles, dihedrals) as a function of time
- Reaction coordinates are defined to track the progress of the reaction
- Bond distances, angles, or dihedrals that change significantly during the reaction
- Collective variables (contact maps, solvent accessible surface area) can capture more complex reaction coordinates
- Transition states are identified as the highest energy points along the reaction coordinate
- Correspond to saddle points on the potential energy surface
- Can be located by scanning the reaction coordinate or using transition path sampling methods
Free Energy Calculations and Kinetics
- Free energy methods (umbrella sampling, metadynamics) can be used to calculate free energy profiles along reaction coordinates
- Umbrella sampling applies biasing potentials to sample high-energy regions and construct the free energy profile
- Metadynamics adds Gaussian potentials to the energy landscape, encouraging the system to explore new regions
- Free energy barriers can be estimated from the free energy profile
- Correspond to the difference in free energy between the reactant and transition state
- Related to the reaction rate through transition state theory
- Transition state theory (TST) can be applied to calculate reaction rates from the free energy barrier and the prefactor
- The prefactor depends on the vibrational frequencies of the reactant and transition state
- TST assumes a quasi-equilibrium between the reactant and transition state and neglects recrossing events
- Committor analysis can be used to identify true transition states
- Launches multiple simulations from putative transition state configurations
- Determines the probability of committing to reactant or product states
- True transition states have a committor probability of 0.5
Mechanistic Insights and Experimental Validation
- Analyzing the sequence of events leading to product formation provides mechanistic insights
- Identifies the role of specific residues, solvent molecules, or cofactors in catalyzing the reaction
- Reveals the order of bond breaking and formation events and the presence of intermediate states
- Comparing simulation results with experimental data validates the simulations and provides a more comprehensive understanding of the reaction
- Kinetic measurements (rate constants, activation energies) can be compared with simulated values
- Spectroscopic observations (IR, Raman, NMR) can be related to structural changes observed in the simulations
- Simulations can guide the design of new experiments to test hypotheses or probe specific aspects of the reaction mechanism
- Suggests mutations or modifications that may affect the reaction rate or selectivity
- Identifies key residues or interactions that can be probed experimentally (mutagenesis, spectroscopy)
Limitations of Molecular Dynamics
Force Field Accuracy and Sampling Limitations
- Force field accuracy is a major limitation of molecular dynamics simulations
- The quality of the simulation depends on the ability of the force field to accurately describe the interatomic interactions
- Force fields are parameterized to reproduce experimental data, but may not capture all the relevant physics
- Sampling limitations arise from the finite simulation time and the challenge of exploring all relevant regions of the potential energy surface
- High energy barriers may prevent the system from accessing important regions of configuration space
- Rare events (reactive trajectories) may not be observed in the limited simulation time
- Enhanced sampling techniques (replica exchange, accelerated molecular dynamics) can help overcome sampling limitations
- Replica exchange simulates multiple copies of the system at different temperatures, allowing for exchanges between replicas
- Accelerated molecular dynamics adds a boost potential to the energy landscape, facilitating the crossing of energy barriers
Quantum Effects and Electronic Structure
- Quantum effects (tunneling, zero-point energy) are not explicitly included in classical molecular dynamics simulations
- Tunneling allows particles to pass through energy barriers, which can be important for reactions involving light atoms (hydrogen)
- Zero-point energy is the lowest possible energy of a quantum system and can affect the relative stability of reactants and products
- The representation of the electronic structure is simplified in classical molecular dynamics
- Electrons are not explicitly modeled, and the potential energy is a function of nuclear positions only
- This may limit the accuracy for systems with significant electronic rearrangements or charge transfer during the reaction
- Ab initio molecular dynamics (AIMD) methods incorporate electronic structure calculations into the molecular dynamics simulation
- The electronic structure is calculated on-the-fly using quantum mechanical methods (density functional theory)
- AIMD can describe bond breaking and formation, but is computationally expensive and limited to small system sizes and short timescales
Finite Size Effects and Statistical Errors
- Finite size effects can influence the results of molecular dynamics simulations
- Simulations of condensed-phase systems (liquids, solids) are limited to small system sizes due to computational cost
- The system size must be sufficiently large to capture the relevant phenomena and avoid artificial boundary effects
- Periodic boundary conditions are used to mimic an infinite system, but can introduce artifacts
- Long-range interactions (electrostatics) must be treated carefully to avoid spurious correlations between periodic images
- Ewald summation methods (particle mesh Ewald) are used to efficiently calculate long-range interactions in periodic systems
- Statistical errors arise from the finite number of trajectories or configurations sampled
- Molecular dynamics simulations are stochastic in nature, and the results can vary between different runs
- Averaging over multiple independent simulations can reduce statistical errors and improve the reliability of the results
- Convergence of the simulations should be carefully assessed
- Monitoring the evolution of thermodynamic properties (energy, temperature) and structural parameters (distances, angles) can indicate whether the system has reached a stable state
- Block averaging or autocorrelation analysis can be used to estimate the statistical uncertainty in the calculated properties
Comparison with Experimental Data
- Comparison with experimental data is essential for validating the accuracy of molecular dynamics simulations
- Kinetic measurements (rate constants, activation energies) provide a direct test of the simulated reaction dynamics
- Structural data (X-ray crystallography, NMR) can be used to assess the quality of the simulated structures and interactions
- Discrepancies between simulations and experiments can arise from various sources
- Inaccuracies in the force field parameters or the treatment of electronic structure
- Insufficient sampling of the relevant configurational space
- Differences in the experimental conditions (temperature, pressure, solvent) compared to the simulated system
- Iterative refinement of the computational methodology based on experimental data can improve the accuracy and predictive power of the simulations
- Force field parameters can be optimized to better reproduce experimental observables
- Enhanced sampling methods can be employed to overcome sampling limitations and explore relevant regions of the potential energy surface
- The simulated system can be modified to more closely match the experimental conditions (e.g., inclusion of explicit solvent molecules or counterions)
- A combination of molecular dynamics simulations and experimental studies provides the most comprehensive understanding of reaction dynamics and mechanisms
- Simulations can provide atomic-level insights and mechanistic hypotheses that can be tested experimentally
- Experiments can guide the development and refinement of computational models, ensuring their accuracy and reliability