Ab initio molecular dynamics goes beyond traditional methods, combining quantum mechanics with classical dynamics. It captures complex molecular behavior, including non-adiabatic effects where electronic and nuclear motions are strongly coupled.
These advanced techniques allow for more accurate simulations of chemical reactions and excited-state dynamics. They're crucial for understanding phenomena like photochemistry and conical intersections, where multiple electronic states interact.
Born-Oppenheimer Approximation and Beyond
Fundamentals of Born-Oppenheimer Approximation
- Born-Oppenheimer approximation separates electronic and nuclear motions in molecular systems
- Assumes electrons move much faster than nuclei due to mass difference
- Allows calculation of electronic structure for fixed nuclear positions
- Simplifies quantum mechanical calculations for molecules
- Breaks down in systems with strong coupling between electronic and nuclear motions
Non-Adiabatic Transitions and Conical Intersections
- Non-adiabatic transitions occur when Born-Oppenheimer approximation fails
- Involve coupling between different electronic states
- Conical intersections represent points where two or more potential energy surfaces meet
- Facilitate rapid transitions between electronic states
- Play crucial roles in photochemical reactions (photosynthesis, vision)
Quantum Nuclear Effects
- Quantum nuclear effects arise from quantum mechanical nature of nuclei
- Include zero-point energy, tunneling, and nuclear delocalization
- Become significant for light atoms (hydrogen) or at low temperatures
- Affect reaction rates and molecular properties
- Require advanced computational methods to accurately model (path integral molecular dynamics)
![Fundamentals of Born-Oppenheimer Approximation, science [CP2K Open Source Molecular Dynamics ]](https://storage.googleapis.com/static.prod.fiveable.me/search-images%2F%22Born-Oppenheimer_approximation_electronic_nuclear_motion_separation_computational_chemistry_image_diagram%22-science%3Adoi_10_1002_anie_201311189_pub.png)
Dynamics Methods for Non-Adiabatic Systems
Surface Hopping and Ehrenfest Dynamics
- Surface hopping simulates non-adiabatic dynamics through discrete transitions between electronic states
- Trajectories evolve on single potential energy surface with probabilistic switches
- Ehrenfest dynamics uses mean-field approach to evolve nuclear coordinates
- Combines multiple electronic states weighted by their populations
- Both methods balance computational efficiency with accuracy for non-adiabatic systems
Semiclassical Dynamics and Fewest Switches Algorithm
- Semiclassical dynamics approximates quantum effects within classical framework
- Incorporates quantum phase information into classical trajectories
- Improves description of interference and tunneling phenomena
- Fewest switches algorithm minimizes number of surface hops in surface hopping simulations
- Ensures energy conservation and improves computational efficiency
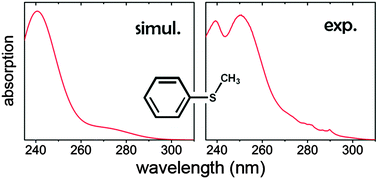
Advanced Ab Initio Molecular Dynamics
Car-Parrinello Molecular Dynamics
- Car-Parrinello molecular dynamics combines electronic structure calculations with classical nuclear dynamics
- Uses fictitious electron dynamics to propagate electronic wavefunctions
- Avoids expensive self-consistent field calculations at each time step
- Enables simulation of large systems for extended time periods
- Balances accuracy of ab initio methods with efficiency of classical molecular dynamics
Quantum Nuclear Effects in Molecular Dynamics
- Path integral molecular dynamics incorporates quantum nuclear effects into simulations
- Represents quantum particles as classical ring polymers
- Captures zero-point energy and tunneling in molecular systems
- Improves accuracy for systems with light atoms or at low temperatures
- Requires increased computational resources compared to classical molecular dynamics
Simulating Non-Adiabatic Transitions and Conical Intersections
- Advanced ab initio molecular dynamics methods model non-adiabatic transitions
- Include multiple electronic states and their couplings
- Capture dynamics near conical intersections
- Enable simulation of photochemical reactions and excited-state dynamics
- Require careful treatment of electronic structure and nuclear dynamics