X-ray crystallography is a powerful technique for revealing the 3D structure of molecules. It uses X-ray diffraction patterns from crystals to map electron density, showing atom positions. This method is crucial for understanding protein structures and functions.
Protein crystallization is a key step, involving careful control of conditions to grow high-quality crystals. Once obtained, these crystals are analyzed to create detailed electron density maps, which are then used to build and refine molecular models.
X-ray Crystallography Principles
Fundamentals of X-ray Diffraction
- X-ray crystallography utilizes the diffraction of X-rays by the regularly spaced atoms in a crystal to determine the three-dimensional structure of molecules
- The diffraction pattern produced when X-rays interact with a crystalline sample is recorded and analyzed to determine the electron density distribution within the crystal
- Fourier transform is applied to the diffraction data to convert it into an electron density map, which reveals the positions of atoms in the molecule
Factors Affecting Structural Determination
- The quality and resolution of the electron density map depend on factors such as crystal size, purity, and the wavelength of the X-rays used
- Molecular models are built by fitting the known amino acid sequence of the protein into the electron density map, followed by refinement to improve the agreement between the model and the experimental data
- X-ray crystallography can determine the structure of proteins, nucleic acids, and other biological macromolecules at atomic resolution, typically in the range of 1-3 Å (angstroms)
- The three-dimensional structures obtained by X-ray crystallography provide insights into the function, interactions, and mechanisms of action of biological molecules (enzymes, receptors)
Protein Crystallization Process

Methods and Techniques
- Protein crystallization is the process of inducing the formation of ordered, repeating arrays of protein molecules in a supersaturated solution
- The most common method for protein crystallization is vapor diffusion, which involves mixing the protein solution with a precipitant solution and allowing the mixture to equilibrate against a reservoir containing a higher concentration of the precipitant
- As water vapor diffuses from the protein drop to the reservoir, the protein concentration increases, leading to supersaturation and potentially crystal nucleation and growth
- Seeding techniques, such as microseeding or macroseeding, can be used to promote crystal growth and improve crystal quality by introducing preformed crystal nuclei into the crystallization drop
Factors Influencing Crystallization Success
- Factors that influence protein crystallization include protein purity, concentration, pH, temperature, and the presence of additives or ligands (metal ions, cofactors)
- The choice of precipitant, such as salts (ammonium sulfate), polymers (polyethylene glycol), or organic solvents (isopropanol), can significantly impact the success of crystallization and the quality of the resulting crystals
- The quality of protein crystals is assessed by their size, morphology, and the resolution of the diffraction pattern they produce when exposed to X-rays
- High-quality crystals are essential for obtaining accurate and high-resolution structural information by X-ray crystallography
Electron Density Maps
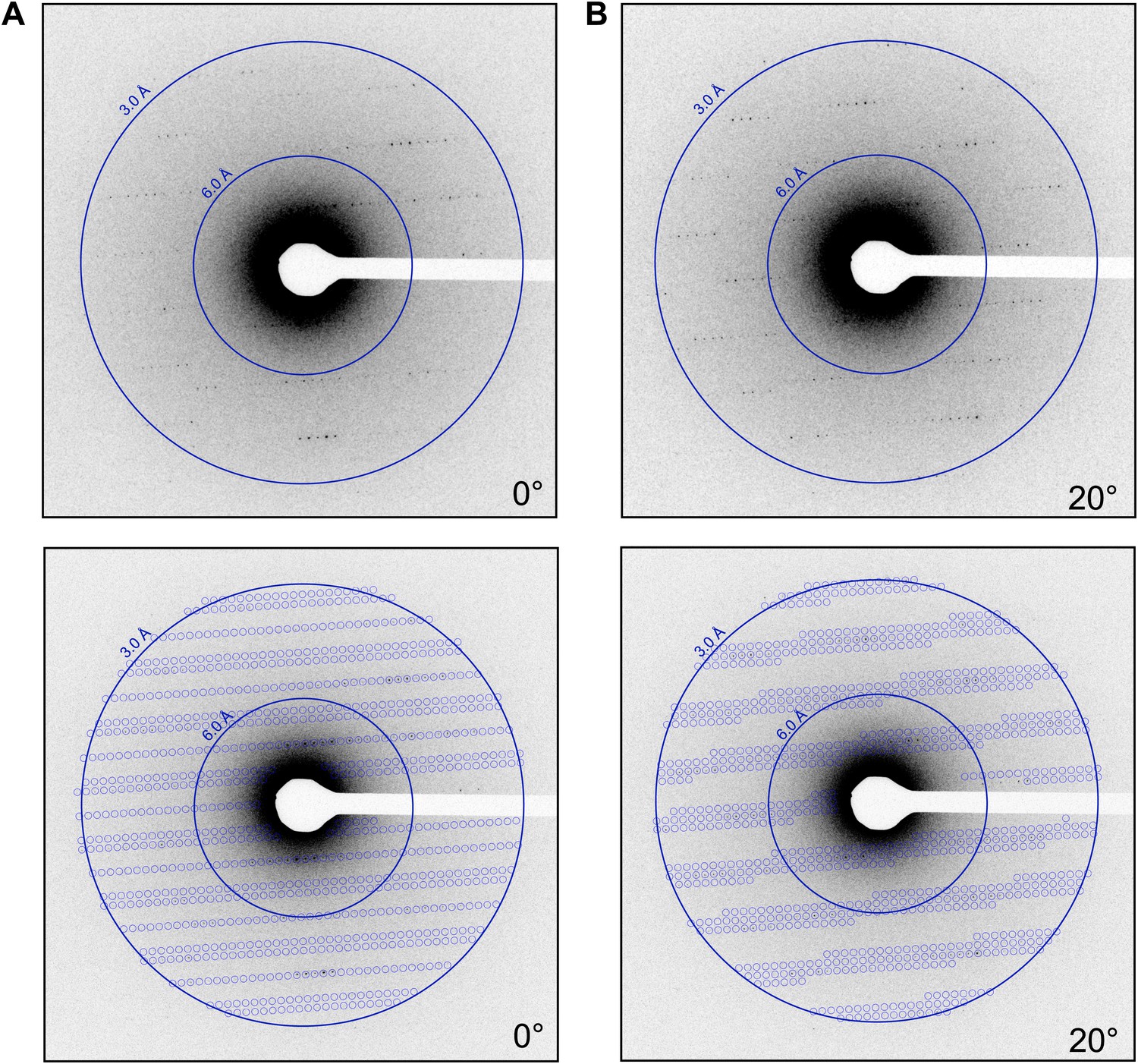
Interpretation of Electron Density
- Electron density maps represent the three-dimensional distribution of electrons in the crystal, which corresponds to the positions of atoms in the molecule
- The electron density at a given point in the map is proportional to the number of electrons present at that location, with higher density indicating the presence of heavier atoms or multiple atoms
- The resolution of the electron density map determines the level of detail visible, with higher resolution maps allowing for the identification of individual atoms and the precise positioning of side chains
- The interpretation of electron density maps involves the recognition of characteristic patterns corresponding to specific amino acid residues (glycine, tryptophan), secondary structures (alpha helices, beta sheets), and other structural features of the molecule
Model Building and Refinement
- Molecular model building is the process of fitting the known amino acid sequence of the protein into the electron density map, using the map as a guide for positioning the atoms
- The initial model is typically built using automated or semi-automated methods, followed by manual adjustments to improve the fit to the electron density
- Model refinement is an iterative process that involves adjusting the atomic positions, B-factors (atomic displacement parameters), and occupancies to minimize the difference between the calculated and observed diffraction data
- Throughout the refinement process, the quality of the model is assessed using various validation tools and statistical measures, such as the R-factor and the Ramachandran plot, to ensure its accuracy and reliability
Applications of X-ray Crystallography
Drug Discovery and Design
- X-ray crystallography plays a crucial role in drug discovery by providing detailed structural information on drug targets, such as enzymes (kinases), receptors (G protein-coupled receptors), and transport proteins (ion channels)
- The three-dimensional structures of target proteins obtained by X-ray crystallography enable the identification of binding sites and the design of molecules that can specifically interact with these sites to modulate protein function
- Structure-based drug design involves the use of protein-ligand complex structures to guide the optimization of lead compounds, improving their potency, selectivity, and pharmacokinetic properties
Protein Engineering and Structure-Function Relationships
- In protein engineering, X-ray crystallography is used to guide the rational design of proteins with enhanced stability, specificity, or catalytic activity
- By comparing the structures of wild-type and mutant proteins, researchers can gain insights into the molecular basis of protein function and the effects of specific mutations on protein stability and activity
- X-ray crystallography is instrumental in elucidating the structure-function relationships of biomolecules, providing a detailed understanding of how the three-dimensional arrangement of atoms determines the biological properties of proteins, nucleic acids, and their complexes
- Comparative structural analysis of related proteins from different organisms or protein families can reveal evolutionary relationships and provide insights into the molecular mechanisms of protein function and regulation (enzyme catalysis, signal transduction)
- The integration of structural information obtained by X-ray crystallography with other experimental data, such as biochemical, biophysical, and computational studies, enables a comprehensive understanding of the structure, dynamics, and function of biological systems at the molecular level