🧬Bioinformatics Unit 4 Review
4.3 Genome annotation
4.3 Genome annotation
Unit & Topic Study Guides
Fundamentals of molecular biology
Bioinformatics: Key Databases and Resources
Sequence alignment algorithms
Genomics and Next-Gen Sequencing
Proteomics & Protein Structure Prediction
Phylogenetics and Evolution Analysis
Gene Expression and Transcriptomics
Machine learning in bioinformatics
Systems Biology & Network Analysis
Structural bioinformatics
Comparative genomics
Genome annotation is a crucial process in bioinformatics that identifies and labels functional elements within DNA sequences. It bridges raw genetic data with biological meaning, using computational algorithms and experimental data to decode the genome's blueprint.
Annotation involves identifying various genomic features like protein-coding genes, non-coding RNAs, and regulatory elements. It employs different prediction methods, functional annotation techniques, and specialized pipelines for prokaryotic and eukaryotic genomes. Challenges include distinguishing pseudogenes and handling alternative splicing.
Overview of genome annotation
- Genome annotation identifies and labels functional elements within genomic sequences, crucial for understanding genetic information
- Combines computational algorithms and experimental data to assign biological meaning to DNA sequences
- Plays a vital role in bioinformatics by bridging raw sequence data with functional genomics and molecular biology
Types of genome features
Protein-coding genes
- Segments of DNA that encode instructions for producing proteins
- Consist of exons (coding regions) and introns (non-coding regions)
- Include start and stop codons, promoter regions, and untranslated regions (UTRs)
- Vary in length and complexity across different organisms (prokaryotes vs eukaryotes)
Non-coding RNA genes
- Genes that produce functional RNA molecules without being translated into proteins
- Include transfer RNAs (tRNAs), ribosomal RNAs (rRNAs), and small nuclear RNAs (snRNAs)
- Regulatory RNAs such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs)
- Play crucial roles in gene regulation, protein synthesis, and cellular processes
Regulatory elements
- DNA sequences that control gene expression and regulation
- Promoters located upstream of genes initiate transcription
- Enhancers and silencers modulate gene expression from distant locations
- Insulators act as boundaries between different regulatory domains
- Transcription factor binding sites allow for specific protein-DNA interactions
Repetitive sequences
- DNA segments that occur multiple times throughout the genome
- Transposable elements can move within the genome (retrotransposons, DNA transposons)
- Tandem repeats include satellite DNA, minisatellites, and microsatellites
- Segmental duplications involve large genomic regions
- Impact genome structure, evolution, and gene regulation
Gene prediction methods
Ab initio prediction
- Computational approach using statistical models to identify genes without prior knowledge
- Relies on intrinsic sequence features such as codon usage and splice site signals
- Employs hidden Markov models (HMMs) or neural networks to predict gene structures
- Effective for well-studied organisms with known sequence patterns
- Limitations in accuracy for complex genomes or newly sequenced species
Homology-based prediction
- Identifies genes by comparing genomic sequences to known genes from related organisms
- Utilizes sequence alignment tools (BLAST, BLAT) to find similarities
- Transfers annotation information from well-characterized genomes to newly sequenced ones
- Effective for conserved genes but may miss novel or rapidly evolving genes
- Requires high-quality reference genomes and comprehensive databases
RNA-seq-based prediction
- Uses transcriptome data to identify expressed genes and their structures
- Aligns RNA-seq reads to the genome to determine exon-intron boundaries
- Reveals alternative splicing events and novel transcripts
- Provides evidence for gene expression levels and tissue-specific variants
- Challenges include distinguishing between noise and low-expression transcripts
Functional annotation
Gene ontology terms
- Standardized vocabulary to describe gene and protein functions across species
- Organized into three main categories molecular function, biological process, cellular component
- Hierarchical structure allows for different levels of specificity
- Enables systematic analysis of gene sets and functional enrichment studies
- Continuously updated by the scientific community to reflect new discoveries
Protein domains
- Distinct functional or structural units within proteins
- Identified using tools like PFAM, PROSITE, or InterPro
- Provide insights into protein function, structure, and evolution
- Can be used to predict protein-protein interactions and enzymatic activities
- Help in classifying proteins into families and superfamilies
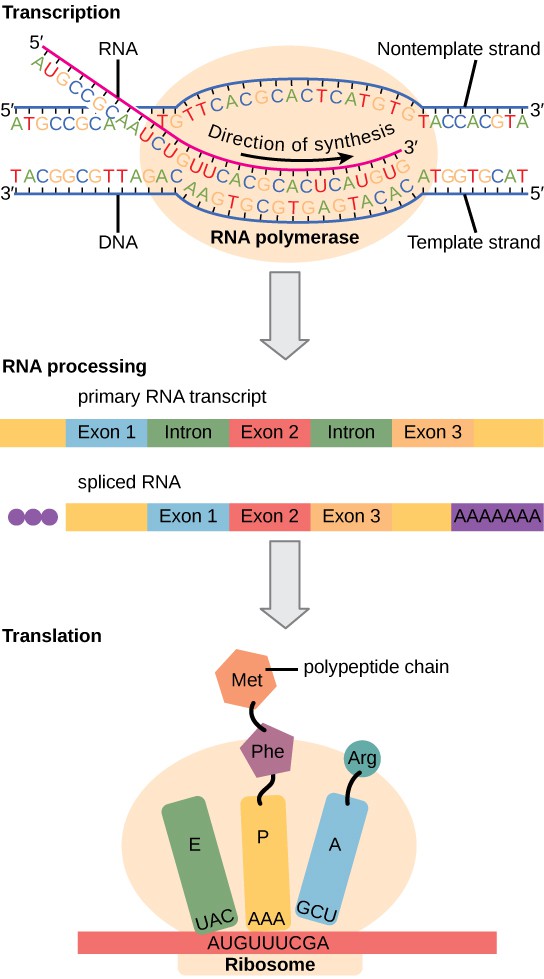
Metabolic pathways
- Series of chemical reactions involved in cellular metabolism
- Annotated using databases like KEGG, MetaCyc, or Reactome
- Link genes and proteins to specific biochemical processes
- Enable the reconstruction of metabolic networks in different organisms
- Useful for understanding cellular functions and identifying potential drug targets
Annotation pipelines
Prokaryotic genome annotation
- Automated pipelines like Prokka or PGAP designed for bacterial and archaeal genomes
- Identify protein-coding genes, tRNAs, rRNAs, and other features
- Utilize databases of known prokaryotic genes and proteins for functional assignment
- Consider unique prokaryotic features (operons, overlapping genes)
- Generally faster and more straightforward than eukaryotic annotation due to simpler genome structure
Eukaryotic genome annotation
- Complex pipelines like MAKER or AUGUSTUS handle intron-exon structures and alternative splicing
- Integrate multiple evidence types (ab initio predictions, RNA-seq data, protein homology)
- Account for repetitive elements and non-coding RNAs
- Often require manual curation to resolve conflicting evidence
- Iterative process involving multiple rounds of refinement and validation
Annotation databases
RefSeq vs GenBank
- RefSeq curated, non-redundant set of reference sequences for genomes, transcripts, and proteins
- GenBank comprehensive public database of all submitted sequences, including raw data
- RefSeq provides a stable reference for each molecule, while GenBank may contain multiple entries
- RefSeq uses accession numbers starting with NC_, NM_, NP_, while GenBank uses various prefixes
- RefSeq undergoes more rigorous curation and quality control compared to GenBank
Ensembl vs UCSC
- Ensembl European-based genome browser and annotation database for vertebrates and other eukaryotes
- UCSC Genome Browser US-based platform for accessing and visualizing genomic data
- Ensembl focuses on automatic annotation and comparative genomics
- UCSC emphasizes manual curation and integration of external data tracks
- Both provide APIs and tools for programmatic access to genomic data and annotations
Challenges in genome annotation
Pseudogenes vs functional genes
- Pseudogenes non-functional gene copies that resemble active genes
- Difficult to distinguish from functional genes due to sequence similarity
- Require integration of multiple evidence types (expression data, evolutionary conservation)
- Can be misannotated as functional genes, leading to overestimation of gene numbers
- Some pseudogenes may retain partial functionality or regulate their parent genes
Alternative splicing
- Process where a single gene can produce multiple mRNA isoforms
- Complicates gene structure prediction and functional annotation
- Requires integration of RNA-seq data to identify different splice variants
- Can lead to underestimation of protein diversity if not properly accounted for
- Varies significantly between species and cell types
Structural variations
- Large-scale genomic differences between individuals or populations
- Include copy number variations, inversions, and translocations
- Can affect gene content, regulation, and function
- Challenging to detect and annotate accurately using short-read sequencing data
- Require specialized algorithms and long-read sequencing technologies for comprehensive annotation
Quality assessment

Annotation completeness
- Evaluates the proportion of the genome that has been successfully annotated
- Uses tools like BUSCO to assess the presence of conserved single-copy orthologs
- Compares gene count and structure to closely related species
- Considers coverage of different feature types (coding genes, ncRNAs, regulatory elements)
- Helps identify areas of the genome that may require further annotation efforts
Consistency checks
- Ensures logical coherence and uniformity across the annotation
- Verifies gene structures for proper start and stop codons, splice sites
- Checks for overlapping features and resolves conflicts
- Compares functional assignments with sequence-based evidence
- Identifies and flags potential annotation errors or inconsistencies
Manual curation
- Expert review and refinement of automated annotations
- Resolves conflicts between different prediction methods or evidence types
- Incorporates domain knowledge and literature-based information
- Improves annotation quality, especially for complex or novel genes
- Time-consuming process, often focused on genes of particular interest or importance
Annotation file formats
GFF vs GTF
- GFF (General Feature Format) flexible, tab-delimited format for describing genomic features
- GTF (Gene Transfer Format) more specialized version of GFF, primarily for gene-centric annotations
- GFF allows for custom feature types and attributes, while GTF has a more rigid structure
- GFF commonly used for a wide range of genomic features, GTF primarily for transcripts and genes
- Both formats support hierarchical relationships between features (gene > transcript > exon)
BED format
- Simple, flexible format for describing genomic intervals
- Contains chromosome, start, end coordinates, and optional additional fields
- Widely used for representing various genomic features and analysis results
- Easily parsed and manipulated by many bioinformatics tools
- Supports visualization in genome browsers and other graphical tools
Annotation visualization tools
Genome browsers
- Web-based or standalone tools for visualizing genomic annotations and data
- Examples include UCSC Genome Browser, Ensembl, JBrowse, and IGV
- Allow users to navigate through genomic regions and view multiple annotation tracks
- Support integration of custom data and annotations
- Facilitate comparative genomics and exploration of genomic context
Circos plots
- Circular visualization tool for displaying genomic data and relationships
- Useful for showing genome-wide patterns and interactions
- Can represent various data types (gene density, synteny, structural variations)
- Highly customizable for creating publication-quality figures
- Particularly effective for visualizing whole-genome comparisons and rearrangements
Reannotation and updates
Improving existing annotations
- Periodic refinement of genome annotations to incorporate new data and knowledge
- Utilizes updated sequence data, improved algorithms, and additional experimental evidence
- Corrects errors and resolves inconsistencies in previous annotations
- Adds newly discovered features and refines existing feature boundaries
- Crucial for maintaining accurate and up-to-date genomic resources
Version control in annotations
- Tracks changes and updates to genome annotations over time
- Assigns version numbers or dates to different annotation releases
- Maintains backward compatibility and allows for reproducibility of analyses
- Provides documentation of changes between versions
- Enables users to choose appropriate annotation versions for their specific needs