🧬Bioinformatics Unit 3 Review
3.6 Gap penalties
3.6 Gap penalties
Unit & Topic Study Guides
Fundamentals of molecular biology
Bioinformatics: Key Databases and Resources
Sequence alignment algorithms
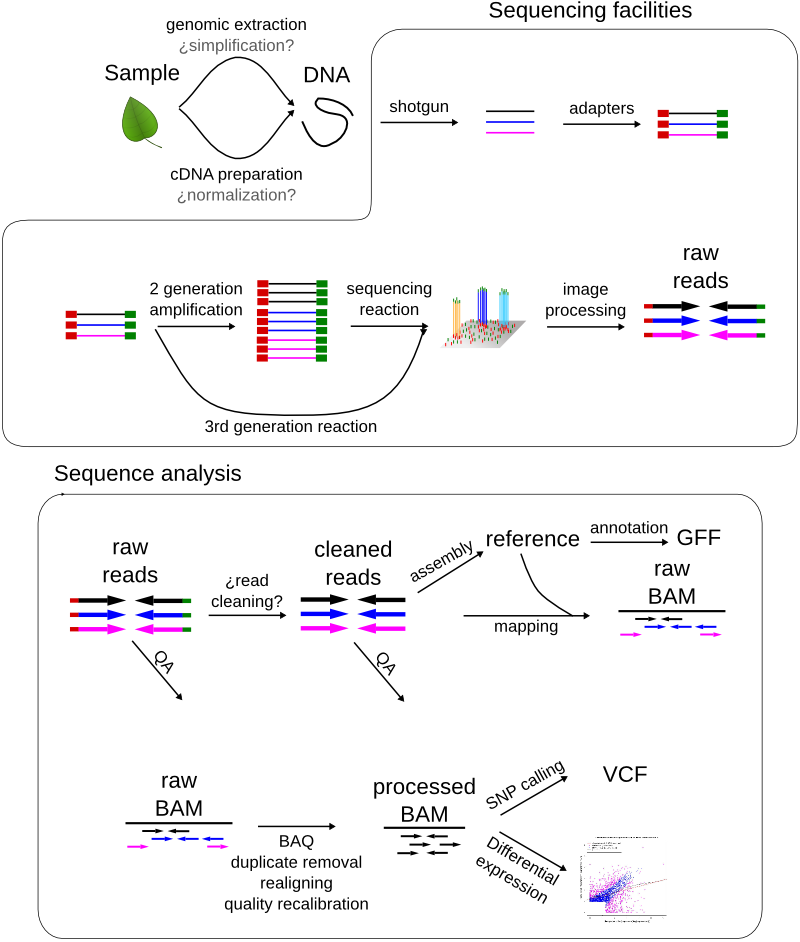
Genomics and Next-Gen Sequencing
Proteomics & Protein Structure Prediction
Phylogenetics and Evolution Analysis
Gene Expression and Transcriptomics
Machine learning in bioinformatics
Systems Biology & Network Analysis
Structural bioinformatics
Comparative genomics
Gap penalties are crucial in bioinformatics for accurate sequence alignment. They account for insertions and deletions in evolutionary processes, helping to compare and analyze genetic sequences more effectively. Different types of gap penalties, such as linear, affine, and convex, offer varying levels of biological realism.
Understanding gap penalties is essential for reconstructing evolutionary relationships between sequences. They influence alignment algorithms, impact computational resources, and affect the balance between sensitivity and specificity in detecting homologies. Advanced models and machine learning approaches continue to refine gap penalty applications in bioinformatics.
Types of gap penalties
- Gap penalties play a crucial role in sequence alignment algorithms used in bioinformatics
- They help account for insertions and deletions that occur during evolutionary processes
- Understanding different types of gap penalties allows for more accurate sequence comparisons and analyses
Linear gap penalties
- Simplest form of gap penalty assigns a fixed cost for each gap regardless of length
- Calculated by multiplying the gap length by a constant penalty value
- Formula: , where k is the constant penalty value
- Advantages include computational simplicity and ease of implementation
- May not accurately reflect biological reality of insertion and deletion events
Affine gap penalties
- Two-part penalty system consisting of gap opening and gap extension penalties
- Reflects biological observation that extending an existing gap is more likely than opening a new one
- Formula: , where o is the opening penalty and e is the extension penalty
- More biologically realistic than linear penalties
- Widely used in popular alignment algorithms (BLAST, CLUSTAL)
Convex gap penalties
- Non-linear penalty system where the cost of extending a gap decreases as the gap length increases
- Accounts for the observation that long gaps are sometimes more likely in certain genomic regions
- Can be implemented using logarithmic or other non-linear functions
- Formula example: , where a and b are constants
- Computationally more complex than linear or affine penalties
Biological basis for gaps
- Gaps in sequence alignments represent evolutionary events that have altered genetic sequences
- Understanding the biological basis for gaps improves the accuracy of alignment algorithms
- Proper gap modeling is essential for reconstructing evolutionary relationships between sequences
Insertions and deletions
- Represent addition or removal of genetic material in a sequence
- Insertions can occur through various mechanisms
- Transposable element insertion
- Duplication events
- Horizontal gene transfer
- Deletions can result from
- DNA repair errors
- Recombination events
- Selection pressure against certain sequences
- Can range from single nucleotides to large genomic regions
Evolutionary significance
- Gaps provide insights into the evolutionary history of sequences
- Help identify conserved regions and functional domains in proteins
- Contribute to understanding of speciation events and phylogenetic relationships
- Can indicate
- Gene fusion or fission events
- Domain shuffling in proteins
- Pseudogene formation
Gap penalty in alignment algorithms
- Gap penalties are integral components of sequence alignment scoring systems
- They influence the placement and length of gaps in the final alignment
- Different alignment algorithms incorporate gap penalties in various ways
Global alignment vs local alignment
- Global alignment (Needleman-Wunsch) aligns entire sequences end-to-end
- Gap penalties applied throughout the alignment
- Suitable for comparing closely related sequences of similar length
- Local alignment (Smith-Waterman) identifies regions of high similarity within sequences
- Gap penalties may be applied differently in high-scoring regions
- Useful for finding conserved domains or motifs in divergent sequences
Smith-Waterman algorithm
- Local alignment algorithm that incorporates gap penalties
- Uses a dynamic programming matrix to find optimal local alignments
- Gap penalties influence the decision to start, extend, or terminate a local alignment
- Can use affine gap penalties to differentiate between gap opening and extension
- Time complexity of where m and n are the lengths of the sequences
Needleman-Wunsch algorithm
- Global alignment algorithm that uses gap penalties in its scoring system
- Employs a dynamic programming approach to find the optimal end-to-end alignment
- Gap penalties affect the choice between introducing a gap or aligning mismatched residues
- Can be modified to use affine gap penalties for more biological realism
- Also has a time complexity of for two sequences of length m and n

Choosing appropriate gap penalties
- Selection of gap penalties significantly impacts alignment quality and biological relevance
- Requires balancing between allowing necessary gaps and preventing excessive gapping
- Often involves empirical testing and domain-specific knowledge
Protein vs DNA sequences
- DNA sequences typically use smaller gap penalties than protein sequences
- Reflects higher frequency of indels in non-coding DNA regions
- Protein gap penalties consider structural implications of insertions/deletions
- Higher penalties in secondary structure elements (alpha-helices, beta-sheets)
- Lower penalties in loop regions or disordered segments
- Codon-aware DNA alignment may use different gap penalties for frame-preserving vs frame-shifting gaps
Impact on alignment accuracy
- Too low gap penalties can lead to over-gapped alignments
- May obscure true homology relationships
- Can artificially inflate sequence similarity scores
- Excessively high gap penalties can force misalignments
- May prevent identification of biologically relevant insertions or deletions
- Can lead to incorrect evolutionary inferences
- Optimal gap penalties often depend on the specific sequences being aligned and the biological question at hand
Empirical determination methods
- Benchmark datasets with known correct alignments (BAliBASE, PREFAB)
- Cross-validation techniques to assess alignment quality
- Parameter sweeping to identify optimal gap penalty values for specific types of sequences
- Use of structural information (for proteins) to guide gap penalty selection
- Machine learning approaches to learn optimal gap penalties from large datasets
Gap opening vs gap extension
- Distinction between gap opening and extension penalties reflects biological realities of insertion/deletion events
- Allows for more nuanced control over gap placement in sequence alignments
Rationale for distinction
- Opening a new gap is generally considered less likely than extending an existing one
- Reflects biological processes such as
- Insertion of transposable elements (often occur as single events)
- Slippage during DNA replication (can lead to extension of existing gaps)
- Helps prevent excessive fragmentation of alignments
- Allows for longer, biologically meaningful gaps when appropriate
Typical penalty values
- Gap opening penalties are usually higher than extension penalties
- Common ratios of opening to extension penalties range from 10:1 to 20:1
- Protein alignment example
- Gap opening penalty: -10 to -15
- Gap extension penalty: -0.5 to -2
- DNA alignment example
- Gap opening penalty: -15 to -20
- Gap extension penalty: -1 to -2
- Values may be adjusted based on sequence characteristics and alignment goals
Gap penalties in multiple sequence alignment
- Multiple sequence alignment (MSA) introduces additional complexities in gap penalty application
- Gap penalties influence both the pairwise alignments and the overall MSA structure
Progressive alignment methods
- Build MSA by sequentially aligning sequences or profiles
- Gap penalties affect each pairwise alignment step
- May use different gap penalties for
- Sequence-sequence alignments
- Sequence-profile alignments
- Profile-profile alignments
- Popular tools (CLUSTAL, MUSCLE) allow user-defined gap penalties
Iterative refinement techniques
- Improve initial MSA through cycles of realignment and optimization
- Gap penalties can be dynamically adjusted during refinement process
- May use position-specific gap penalties based on conservation patterns
- Techniques to prevent gap propagation errors
- Gap-open penalty increase in highly gapped regions
- Anchoring of confidently aligned segments
Computational considerations
- Gap penalties significantly impact the computational resources required for sequence alignment
- Understanding these impacts is crucial for designing efficient alignment algorithms
Time complexity impact
- Introducing gaps increases the search space for optimal alignments
- Affine gap penalties require additional dynamic programming matrices
- Increases time complexity by a constant factor
- For pairwise alignment, time complexity remains but with larger constant factors
- In multiple sequence alignment, gap penalties can significantly increase runtime
- Especially for iterative refinement methods
Space complexity impact
- Additional memory required to store gap-related information in alignment matrices
- Affine gap penalties typically require 2-3 times more memory than simple linear penalties
- Space-saving techniques (e.g., Hirschberg's algorithm) can be adapted for gap penalty calculations
- Trade-offs between time and space complexity when implementing gap penalties in alignment algorithms
Gap penalties in profile-based methods
- Profile-based methods use position-specific information to improve alignment accuracy
- Incorporation of gap penalties in these methods allows for more sophisticated alignment strategies
Position-specific gap penalties
- Vary gap penalties based on the characteristics of specific positions in a sequence or profile
- Can be derived from
- Conservation patterns in multiple sequence alignments
- Known structural features of proteins
- Evolutionary information from phylogenetic analyses
- Allow for lower gap penalties in variable regions and higher penalties in conserved areas
- Implemented in advanced alignment tools (PSI-BLAST, MAFFT)
Profile hidden Markov models
- Probabilistic models that incorporate position-specific gap probabilities
- Transition probabilities between match, insertion, and deletion states represent implicit gap penalties
- Training of HMMs on multiple sequence alignments can learn optimal gap parameters
- Used in homology detection and alignment tools (HMMER, SAM)
- Allow for sophisticated modeling of insertion/deletion patterns in protein families
Limitations and challenges
- While gap penalties improve alignment accuracy, they also introduce certain limitations and challenges
- Understanding these issues is crucial for interpreting alignment results and developing improved methods
Overextension of gaps
- Tendency for alignment algorithms to create longer gaps than biologically appropriate
- Can occur when gap extension penalties are too low relative to mismatch penalties
- May lead to
- Artificially inflated sequence similarity scores
- Incorrect inference of insertion/deletion events
- Mitigation strategies include
- Careful tuning of gap penalty parameters
- Use of local alignment techniques
- Implementation of length-dependent gap penalties
Balancing sensitivity and specificity
- Gap penalties affect the trade-off between detecting true homologies and avoiding false positives
- Lower gap penalties increase sensitivity but may lead to spurious alignments
- Higher gap penalties improve specificity but may miss biologically relevant similarities
- Challenges in finding optimal balance for diverse sequence types and evolutionary distances
- Approaches to address this issue
- Use of adaptive gap penalties based on sequence composition
- Incorporation of additional biological information (structure, function) in alignment scoring
- Development of statistical models to assess alignment significance
Advanced gap penalty models
- Ongoing research in bioinformatics aims to develop more sophisticated gap penalty models
- These advanced models seek to better capture the complexities of biological sequence evolution
Context-dependent gap penalties
- Adjust gap penalties based on local sequence context
- Consider factors such as
- Amino acid composition surrounding potential gap sites
- Secondary structure predictions for protein sequences
- DNA motifs or regulatory elements in genomic sequences
- Implementation examples
- Reduced gap penalties in hydrophilic regions of proteins
- Increased penalties for breaking conserved DNA motifs
- Challenges include increased computational complexity and potential overfitting
Machine learning approaches
- Use of artificial intelligence techniques to learn optimal gap penalty schemes
- Can incorporate diverse features beyond simple sequence information
- Evolutionary conservation scores
- Structural data for proteins
- Functional annotations
- Methods include
- Neural networks for predicting gap locations
- Support vector machines for optimizing gap penalty parameters
- Deep learning models for end-to-end alignment optimization
- Potential to capture complex, non-linear relationships in gap formation patterns
- Requires large, high-quality training datasets of known alignments